糖原累积病
更新时间:2026-04-28
简介
病因
(一) 发病原因
糖原贮积病为常染色体隐性遗传,磷酸化酶激酶缺乏型则是X-性连锁遗传。
(二) 发病机制
糖原常作为备用能量主要存在于肝和肌肉中,正常的肝和肌肉分别含有约4%和2%糖原。是以葡萄糖为基本单位构成的多元糖,分子量大。进入人体内的葡萄糖分别经过葡萄糖激酶,葡糖磷酸变位酶和尿苷二磷酸葡糖焦磷酸化酶的催化作用,转化为尿苷二磷酸葡萄糖(UDPG),之后在糖原合成酶的作用下将以α-1,4-糖苷键的形式连接成一个长链;每隔3~5个葡萄糖残基的由1,4位连接的葡萄糖被分支酶的作用转化为1,6位连接,依次下去,因为形成分支最终构成树状结构的大分子糖原,其分子量达数百万以上。糖原合成和分解代谢中所必需的各种酶至少有8种,GSD发病是由于患者缺乏上述任一酶的缺陷使糖原合成或分解发生障碍,导致糖原沉积于组织中而致病,由于酶缺陷的种类不同,临床表现不同。
Ⅰ型:由于细胞内一种可降解葡糖胺聚酶(GAG)的酶缺乏或障碍所致的。
Ⅱ型:是艾杜糖醛酸-2-硫酸酯酶缺乏。
Ⅲ型:酶的缺乏各亚型不同。ⅢA型为硫酸酰胺酶(旧名称类肝素-N-硫酸酯酶)缺乏,ⅢB为α-N-乙酰葡糖胺酶缺乏,ⅢC为N-乙酰基转移酶缺乏,ⅢD为葡糖胺-6-硫酸酯酶缺乏
Ⅳ型(Morquio氏病),有两个亚型。其病因为ⅣA为半乳糖-6-硫酸酯酶缺乏,ⅣB为β-D半乳糖酶缺乏。
Ⅴ型:为黏多糖贮积症Ⅰ型的Seheie型,由于胺聚酶(GAG)的酶缺乏或障碍所致的。
Ⅵ型:为N-乙酰半乳糖胺-4-硫酸酯酶缺乏,临床上分重型和轻型。
Ⅶ型:是β-D-葡糖醛酸酶缺乏。
Ⅷ型:是由于N-乙酰氨基葡糖-6-硫酸酯酶缺乏。
症状体征
一 临床症状:出生即可发病,成年之后,轻病者的患者会有所好转。以肝脏病为主的Ⅰ型最为常见。
患儿出生时就会有肝脏肿大的症状。新生儿肝肿大不明显,而不被注意。1岁左右逐渐见肝脏肿大,甚至占据整个腹腔。
低血糖:多于1岁以内出现,随着年龄增长,会出现明显低血糖症状,例如软弱呕吐、无力、出汗、惊厥和昏迷,
生长发育迟缓:反复发作的低血糖会影响患者的智力发育以及身体发育,表现为智能低下,患者肥胖体、个子矮小、皮肤暗淡,颜色多为淡黄色,肌肉发育差,较常见下肢无力的症状。
酮症酸中毒:是小儿死亡的主要原因。多数病人在发生意识障碍前数天有多尿、烦渴多饮和乏力,随后出现食欲减退、恶心、呕吐,常伴头痛、嗜睡、烦躁、呼吸深快,呼气中有烂苹果味(丙酮)是其典型发作时候的特点。随着病情进一步发展,出现严重失水,尿量减少,皮肤弹性差,眼球下陷,脉细速,血压下降。至晚期时各种反射迟钝甚至消失,嗜睡以至昏迷。
二 分型
1.Ⅰ型(VonGeirk病):最常见。主要表现低血糖、肝大、酸中毒、高脂血症高尿酸血症高乳酸血症、凝血功能障碍、发育迟缓等临床症状。
2.Ⅱ型:较为少见。其中A型的病情较重。患者全部为男性,多于2~6岁起病。临床表现与Hurler综合征相似,但出现时间较晚,进展较缓慢。B型患者病情较轻,有的听力和角膜可均正常,亦无骨骼畸形。
3.Ⅲ型:极为少见。虽然本型可有4种不同的酶缺乏,但其临床表现非常相似,主要为进行性的智力减退,其中以黏多糖贮积症ⅢA型的临床进展较快。
4.Ⅳ型(Morquio氏病):有两个亚型。面容及智力正常。学步较晚,行走时步态蹒跚不稳。短颈、耸肩。出牙时间较晚,牙列不整齐,牙齿缺乏光泽。角膜混浊可早在儿童期开始出现。听力呈进行性损害。
5.Ⅴ型:现认为该型即为黏多糖贮积症Ⅰ型的Seheie型,与Hrular综合征不同之处表现为无严重的角膜混浊,且混浊为周边性,患者智力正常,身材正常或稍矮,寿命基本正常,但有多毛,关节强直。背柱、头颅X线示仅有轻微改变。
6.Ⅵ型(Maroteaux-Lamy综合征)。临床上分重型和轻型。极为罕见。临床表现与黏多糖贮积症Ⅰ型相似,但患者的智力正常。一般从2~3岁开始出现生长迟缓。颅骨缝闭合较早,可出现脑积水,并引起颅高压症状和痉挛性偏瘫。角膜混浊出现较早,有进行性听力损害,严重者有失明和耳聋。
7.Ⅶ型:极罕见。特殊面容在出生后不久即开始逐渐出现。一般智力正常,角膜混浊及听力损害较常见。多有肝脾肿大,通常不累及心脏,无腹外疝。上肢较短,骨骼发育不良,可有鸡胸、膝外翻等骨骼畸形。
8.Ⅷ型:临床表现有黏多糖病Ⅲ型和Ⅳ型的共同特征,有侏儒,智能落后,脏器受累和骨骼畸形,无角膜混浊。
三 诊断
在新生儿和婴幼儿下频发低血糖抽搐和神智不清,喂食或注射葡萄糖后即可恢复;在出现低血糖的同时有呼吸深快的酸中毒症状,这是诊断糖原累积病的重要临床线索。体征可见肝脏肿大、右上腹隆起。实验室检查应包括血糖、血酮体、乳酸、血脂和尿酸(禁食和餐后)的动态变化。
糖原累积病吃什么好
饮食治疗是一种对症治疗,使用高蛋白、高葡萄糖饮食,少量多次喂养,便可达到维持血糖正常水平防止低血糖症发生的目的。
自从Folkman等在1972年首次证实全静脉营养(TPN)疗法可以纠正本病的异常生化改变和改善临床症状以来,一种日间多次少量进食和夜间应用鼻饲管持续点滴高碳水化合物液的治疗方案曾被广泛使用。通常以维持血糖水平在4~5mmol/L为宜。这种治疗方法不仅可以消除临床症状,并且还使患儿获得
鉴别诊断
1.各类型间的鉴别诊断:利用糖代谢功能实验进行各项指标检查。Ⅰ型血糖累积病的确诊应以肝组织的定量和葡萄糖-6-磷酸酶活性测定为依据,其他各型亦依酶学检查确诊。
2.本病需与糖尿病相鉴别:糖尿病患者也有酸中毒,低血糖的症状,但糖尿病患者有典型的症状是“三多一少”,即多饮、多尿、多食及消瘦,根据次症状可鉴别。
3.本病需与肥厚型心肌病相鉴别:由某些基因突变引起的糖原贮积型心肌病可以模仿肥厚型心肌病,可利用电生理学现象区别。但该病有电生理学异常,而肥厚型心肌病后者没有。
4.肝性糖原累积病有低血糖者应与其他原因引起低血糖的疾病鉴别,可根据有无酮中毒和血乳酸水平来鉴别,其他疾病引起的低血糖一般均无前述实验室检查。
5.肌肉单独受累的糖原累积病(如V型)则应与其他代谢性肌病鉴别,如线粒体肌病、进行性肌营养不良、近端肌紧张性肌病等。肌肉活检有助于作出鉴别诊断,由糖原累积病引起者肌细胞中有糖原堆积。
并发症
腹部膨隆,肝脾肿大,多有腹股沟疝或脐疝,可有腹泻或便秘。
常伴有高尿酸血症,这是由于患儿嘌呤合成代谢亢进所致。
可出现严重低血糖、酸中毒、呼吸困难,惊厥,鼻出血,骨质疏松,胰腺炎和肝腺瘤(或腺癌)等。
可并发乳酸血症、酮尿,高脂血症,感染,严重的可死于酸中毒或感染等疾病。
预防保健
治疗用药
遗传咨询
对于有家族病史的要遗传咨询可以帮助患病成人进行选择性生育。
婚前检查:包括详细询问男女双方及其家庭成员的健康状况既往病史及医治情况,尤其是有无先天畸形,遗传病史和近亲婚配史。应进行家系调查、血型检查染色体检查或基因诊断,以检出携带者;
产前咨询:
孕妇:孕期用药要经医生的指导。做好产前的检查。患者生子女必须进行产前诊断。
在妊娠期穿刺羊水进行检查:①在羊水中可发现黏多糖增多(但在妊娠16周以前并无诊断意义);②羊水细胞培养后,对成维细胞进行分析,可做出早期诊断,从而早期终止妊娠。
保健品查询热门体检套餐





大家都在看
- 乳腺癌
乳腺癌(mammarycancer)是女性最常见的恶性肿瘤之一,发病率占全身各种...[详细]
- 男性不育症
男性不育症是指由于男性因素引起的不育。一般把婚后同居2年以上未采取任何避孕措施而...[详细]
- 不孕症

不孕症(infertility)系指凡婚后夫妇有正常的性生活、未避孕、同居2年而...[详细]
- 早泄
早泄是指阴茎插入阴道后,在女性尚未达到性高潮,而男性的性交时间短于2分钟,提早射...[详细]
- 胃癌

胃癌是我国最常见的恶性肿瘤之一,在我国其发病率居各类肿瘤的首位,每年约有17万人...[详细]
- 前列腺炎
前列腺炎是指前列腺特异性和非特异感染所致的急慢性炎症,从而引起的全身或局部症状。...[详细]
- 流产
流产(abortion)为妇科常见疾病,如处理不当或处理不及时,可能遗留生殖器官...[详细]
- 乙肝
乙型病毒性肝炎是由乙型肝炎病毒(HBV)引起的一种世界性疾病。发展中国家发病率高...[详细]
- 颈椎病
颈椎病又称颈椎综合征,是颈椎骨关节炎、增生性颈椎炎、颈神经根综合征、颈椎间盘脱出...[详细]
- 心绞痛
心绞痛(anginapectoris)是冠状动脉供血不足,心肌急剧的、暂时缺血与...[详细]
最新
- 肿瘤

肿瘤(tumour)是指机体在各种致瘤因子作用下,局部组织细胞增生所形成的新生物...[详细]
- 心脏病

心脏病是一类比较常见的循环系统疾病。循环系统由心脏、血管和调节血液循环的神经体液...[详细]
- 脑溢血
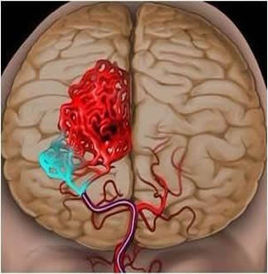
脑出血是指非外伤性脑实质内血管破裂引起的出血,占全部脑卒中的20%~30%,发生...[详细]
- 内分泌性高血压
内分泌组织增生或肿瘤所致的多种内分泌疾病,由于其相应激素如醛固酮、儿茶酚胺、皮质...[详细]
- 寒痹
寒痹是怎么回事?寒痹,病名。一名痛痹、骨痹。指寒邪偏重的痹证。《灵枢•贼风》:“...[详细]
- 小儿46-XY单纯性腺发育不全综合征
46-XY单纯性腺发育不全综合征(simple46,XYgonadaldigen...[详细]
- 小儿情感交叉擦腿综合征

情感交叉擦腿综合征(masturbationsyndrome)是一个病因不明,治...[详细]
- 小儿先天性肌强直综合征

肌强直综合征(myotonicsyndrome)是一组较为少见的遗传性疾病,包括...[详细]
- 新生儿单纯疱疹病毒感染
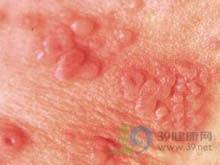
新生儿单纯疱疹病毒感染(herpessimplexviralinfectiono...[详细]
- 小儿猫叫综合征

猫叫综合征(catscrysyndrome)是由于第5号染色体短臂缺失(5p缺失...[详细]

