苍白球黑质红核色素变性
更新时间:2026-04-28
苍白球黑质红核色素变性也称Hallervorden-Spatz病,为儿童晚期和青少年期遗传代谢性疾病。主要累及锥体外系统,也是罕见的与铁元素代谢障碍有关的神经变性病,国内已有少数尸检的报道。本病由Hallervorden和Spatz(1922)首先报道,以后就以两人名命此病。
本病为常染色体隐性遗传。由于铁盐沉积于双侧苍白球、黑质网状部,或甚至红核,导致神经变性,伴有神经元脱失和胶质化。主要临床表现为儿童和青少年中缓慢进展的强直、少动、肌张力障碍、锥体束征、痴呆及色素性视网膜炎,并可有视盘萎缩。
简介
病因
(一)发病原因
病因尚不完全清楚,一般认为是常染色体隐性遗传,Taylor用DNA连锁研究发现,该病致病基因位于染色体20p12.3~p13区。
(二)发病机制
发病机制尚不清楚。无明确的特异性生化异常。双侧基底核铁沉积但不伴有血清铁含量异常或铁代谢紊乱,脑脊液、血清及身体其他组织铁含量正常,铁蛋白、转铁蛋白浓度正常,提示可能患者脑组织内铁代谢异常。
不过有报道在静脉给予标记的枸橼酸二价铁后,基底核区域放射性铁吸收过度。铁沉积的意义难以定论,其他变性性疾病在某种程度上也有基底核铁沉积,例如帕金森病和纹状体-黑质变性,铁沉积是正常人的2~3倍,因此,铁含量增高并非本病特异性表现,不能作为诊断本病的依据。
有报道认为由脂质代谢的异常,氧化应激反应使黑质、苍白球中大量存积的铁(Fe2 )从O-2、H2O2等中得到电子,Fe2 变成Fe3 。这些游离的自由基和Fe3 ,导致细胞的死亡,髓鞘损伤。在Hallervorden-Spats病中铁引起的氧化应激起重要作用。在帕金森病和其他变性病中也有类似的发病机制,但为什么类似发病机制在同一基底核损伤中产生不同疾病,并不清楚。
神经病理有特征性改变:苍白球、黑质(特别是前部分)和红核有深棕色色素沉着;颗粒状和不定形的铁、钙混合沉积物附着在小血管壁上或游离于组织中;大多数受累组织神经细胞变性并大量消失,神经纤维脱髓鞘样改变,神经突触变性,神经胶质细胞轻度增生,脑干神经细胞及小脑齿状核细胞亦可累及;有时可发现黑质内存在神经元纤维缠结及Lewy体。
另一个特点是有肿胀的轴突片断存在,这与神经轴突营养不良的病理改变相似,为此,一些神经病理学者认为Hallervden-Spatz病是一种少年型神经轴突营养不良病。但因铁沉积在后一种疾病不明显,这一观点未得到一致承认。
症状体征
1.分型 可分为儿童型和成人型,儿童型多见。
儿童型多于6~12岁起病。Dooling等(1974)复习了本病有尸解的病例中的临床材料。57%(24例)在10岁前发病,81%(34例)在15岁之前发病,仅7%(3例)在22岁后发病。半数病例有家族史。病程10年左右,多数在20~30岁死于并发症。
成人型又称为晚发型。在55岁左右发病,个别在30岁后发病。常有家族史。类似于帕金森病表现,强直少动,静止性震颤,易跌倒,发音缓慢,声音低沉,小步。美多巴治疗效果不明显。少数患者怕光、吞咽不便、大小便失禁、智能减退,甚至痴呆。病程多达10余年。起病后10~20年仍能行走。
2.临床表现 变异很大,双下肢痉挛性瘫、肌张力障碍和肌强直是本病最突出的特点。主要表现为缓慢进展的锥体外系症状,首先出现的是下肢肌强直、肌张力障碍及舞蹈-手足徐动等症状。早期即可有锥体束征,出现痉挛性瘫、腱反射亢进及Babinski征等;逐渐进展累及上肢、面部及延髓肌;有些病人出现舌肌张力障碍、眼睑痉挛或身体背屈成弓形,引起吞咽困难,口齿不清。晚期患者不能起床,多数在起病10年内因并发症死亡。
3.文献也有原发性视神经萎缩的少数病例报道,部分患者可有精神症状,多数患者出现智力下降及衰退,以及共济失调、痫性发作等。部分患者家族中可有手足徐动症、震颤麻痹或肌张力障碍的病人。
根据青少年期出现进行性锥体外系症状,肌张力障碍、肌强直和双下肢痉挛性瘫等,MRI检查T2WI显示典型“虎眼征”,可以诊断本病。
阳性家族、骨髓巨噬细胞中或周围血淋巴细胞中分辨到海蓝色组织细胞或59Fe标记的铁盐的SPECT显像在双侧基底核区有放射性聚积及消失缓慢,则可证实本病。
苍白球黑质红核色素变性吃什么好
暂无相关饮食资料
鉴别诊断
须与可引起锥体外系症状的变性病和代谢性疾病鉴别。
1.青少年型震颤麻痹(juvenile paralysis agitans) Ramsay Hunt(1917)报道本病,特征与成人型帕金森病相似,大多数病人散发,Van Bogaert曾报道家族型病例,尸检发现豆状核萎缩和黑质、苍白球细胞缺失。表现精神症状不明显,进展缓慢,起病10~20年仍能行走,可资鉴别。
2.Chediak-Higashi病 出现神经系统症状时近一半病人表现智能发育障碍、痫性发作、小脑性共济失调及帕金森综合征等,慢性多发性神经病是主要表现。
3.早发型Huntington舞蹈病 5~14岁或早至1~4岁发病,有家族史。近5%的Huntington舞蹈病是青少年型,出现智能发育迟滞,言语缓慢、肢体肌强直、小步态、肢体和躯干屈曲性张力增高等,可出现舞蹈-手足徐动症,偶有Babinski征。有些青少年型病人无肌强直,痫性发作成年型多见。可出现行为异常、违拗症和姿势紧张,以及肌阵挛和小脑性共济失调等。最终病人言语不能,张口、肢体屈曲、手握拳状和躯干扭转状态等。
4.去髓鞘状态(status dysmyelinatus) 是Vogt提出的尚不确定的疾病,豆状核有髓纤维和神经细胞消失。表现锥体外系肌强直,逐渐出现手足徐动症,最终肢体无力或痉挛导致屈曲畸形,常误诊帕金森病。
5.Lafora体病(Lafora-boby) 可出现肌强直、肌震挛、运动不能和震颤,可有痫性发作和痴呆等。
6.Leigh病 临床少见,在儿童后期或成年时逐渐出现锥体外系肌强直,CT显示壳核空洞形成。
7.本病尚需与肝豆状核变性、重金属中毒、脑炎后遗症,以及Segawa型左旋多巴反应性肌张力障碍(Segawa type of L-dopa-responsive dystonia)、齿状核红核苍白球路易体萎缩(dentatorubropallidoluysian atrophy,DRPLA)等鉴别,神经系统症状均类似Wilson病,但无肝脏受累或铜代谢障碍证据。本组疾病很多都有肌痉挛及锥体外系体征如舞蹈-手足徐动、肌张力障碍等,需综合临床和辅助检查资料加以鉴别。
并发症
可合并有原发性视神经萎缩、精神症状、智力下降及衰退、共济失调、痫性发作等。
预防保健
治疗用药
预防:苍白球黑质红核色素变性是一种神经变性疾病,有遗传背景。预防措施包括避免近亲结婚,推行遗传咨询、携带者基因检测及产前诊断和选择性人工流产等,防止患儿出生。早期诊断、早期治疗、加强临床护理,对改善患者的生活质量有重要意义。
热门体检套餐





大家都在看
- 乳腺癌
乳腺癌(mammarycancer)是女性最常见的恶性肿瘤之一,发病率占全身各种...[详细]
- 男性不育症
男性不育症是指由于男性因素引起的不育。一般把婚后同居2年以上未采取任何避孕措施而...[详细]
- 不孕症

不孕症(infertility)系指凡婚后夫妇有正常的性生活、未避孕、同居2年而...[详细]
- 早泄
早泄是指阴茎插入阴道后,在女性尚未达到性高潮,而男性的性交时间短于2分钟,提早射...[详细]
- 胃癌

胃癌是我国最常见的恶性肿瘤之一,在我国其发病率居各类肿瘤的首位,每年约有17万人...[详细]
- 前列腺炎
前列腺炎是指前列腺特异性和非特异感染所致的急慢性炎症,从而引起的全身或局部症状。...[详细]
- 流产
流产(abortion)为妇科常见疾病,如处理不当或处理不及时,可能遗留生殖器官...[详细]
- 乙肝
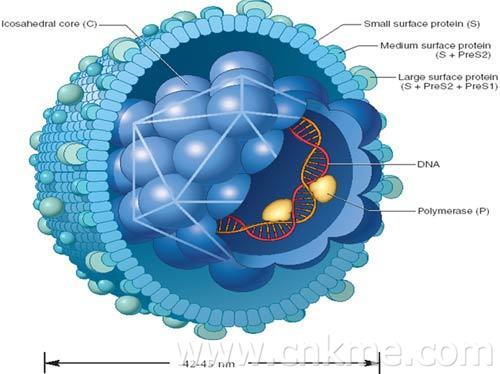
乙型病毒性肝炎是由乙型肝炎病毒(HBV)引起的一种世界性疾病。发展中国家发病率高...[详细]
- 颈椎病
颈椎病又称颈椎综合征,是颈椎骨关节炎、增生性颈椎炎、颈神经根综合征、颈椎间盘脱出...[详细]
- 心绞痛
心绞痛(anginapectoris)是冠状动脉供血不足,心肌急剧的、暂时缺血与...[详细]
最新
- 肿瘤

肿瘤(tumour)是指机体在各种致瘤因子作用下,局部组织细胞增生所形成的新生物...[详细]
- 心脏病

心脏病是一类比较常见的循环系统疾病。循环系统由心脏、血管和调节血液循环的神经体液...[详细]
- 脑溢血
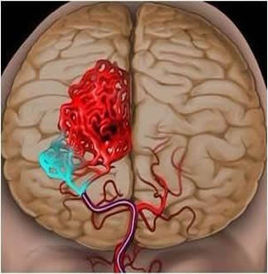
脑出血是指非外伤性脑实质内血管破裂引起的出血,占全部脑卒中的20%~30%,发生...[详细]
- 内分泌性高血压
内分泌组织增生或肿瘤所致的多种内分泌疾病,由于其相应激素如醛固酮、儿茶酚胺、皮质...[详细]
- 寒痹
寒痹是怎么回事?寒痹,病名。一名痛痹、骨痹。指寒邪偏重的痹证。《灵枢•贼风》:“...[详细]
- 小儿46-XY单纯性腺发育不全综合征
46-XY单纯性腺发育不全综合征(simple46,XYgonadaldigen...[详细]
- 小儿情感交叉擦腿综合征

情感交叉擦腿综合征(masturbationsyndrome)是一个病因不明,治...[详细]
- 小儿先天性肌强直综合征

肌强直综合征(myotonicsyndrome)是一组较为少见的遗传性疾病,包括...[详细]
- 新生儿单纯疱疹病毒感染
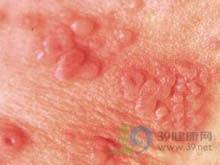
新生儿单纯疱疹病毒感染(herpessimplexviralinfectiono...[详细]
- 小儿猫叫综合征

猫叫综合征(catscrysyndrome)是由于第5号染色体短臂缺失(5p缺失...[详细]

