小儿范科尼综合征
更新时间:2026-04-28
简介
病因
本征可因多种遗传性或获得性疾病引起。年长儿多继发于免疫性疾病、毒物或药物中毒以及各种肾脏病。
导致本征的共同机制可能有二:一是肾小管膜的完整性改变致发生泄漏而不能有效地重吸收多种溶质;二是肾小管细胞内代谢改变,不能产生足够的能量以维持物质的转运。这两者有时不可截然分开,因为维持细胞间的完整性和保持小管上皮需要细胞内的能量。
1.原发性(原因不明或无全身性疾病) 包括遗传性常染色体显性(AD)、常染色体隐性(AR)、X连锁隐性(XLR)、散发性、特殊型(即刷状缘缺失型)。原发性Fanconi 综合征又分为:婴儿型、成人型以及刷状缘缺失型叁种类型。
2.继发性(症状型) 又包括继发于遗传性疾病与继发于后天获得性疾病。前者包括:胱氨酸储积病、酪氨酸血症Ⅰ型、糖原贮积病Ⅰ型、半乳糖血症、遗传性果糖不耐受、细胞色素C 氧化酶缺乏症、Wilson 病、Lowe 综合征、遗传性成骨不全、Alport 综合征、先天性肾病综合征、维生素D 依赖性佝偻病等;后者包括:肾病综合征、移植肾、急慢性间质性肾炎、多发性骨髓瘤肾病、舍格伦综合征、肾淀粉样变性、重金属中毒、药物(过期四环素、氨基糖类抗生素、6-巯基嘌呤、顺铂等)引起的肾损害、低钾性肾病、甲状旁腺功能亢进以及肿瘤相关性肾病等。
幼儿儿童大多同遗传有关,成人则多继发于免疫病、金属中毒或肾脏病。
(二)发病机制
1.发病机制 本病发病机制尚未完全清楚,有以下几种可能:
(1)内流缺陷:管腔内向组织内流减少,见于刷状缘缺失型。
(2)细胞内回漏到肾小管腔增加:如马来酸中毒型。
(3)回流减少:通过基底侧细胞膜回流减少,致细胞内物质堆积;影响回吸收,如Fanconi-Bickel综合征。
(4)灌注增加:从血液向细胞灌注增多,通具体胞紧密保持处反流管腔增多,如细胞色素氧化酶短少型。肾小管膜的输送非常在病理组织学查抄中未见独特点表现。有测验测验提示本征的细胞内活性的转运功能不满是由于磷酸盐耗竭,引起细胞内腺嘌呤核苷酸降解,因而发生消耗。本症的病理心理学改变见图。
2.病理与病理生理 随着分子生物学的研究进展,已认识到FA的发生是一个复杂的病理生理过程,DNA损伤识别或修复缺陷是FA发生的关键,由于DNA的异常启动了相关的病理机制。
(1)DNA交联修复缺陷:FA细胞对能产生链内和链间交联的双功能交联剂(如DEB、MMC、氮芥、环磷酰胺、顺铂等)敏感。DEB和MMC诱导的链内交联修复使DNA链切开,双链DNA同时损伤则没有可用的模板进行修复,需通过非同源末端连接(NHEJ)修复。在FA细胞,非同源重组修复的保真性下降,导致细胞缺陷。相反,通过姊妹染色单体互换的同源重组在FA没有缺陷。除了NHEJ异常,DNA损伤的识别也是受损的,使FA细胞在复制完成以后阻滞在G2期检查点。
(2)FA细胞对氧的超敏性:一种理论认为,FA细胞是被蓄积的氧自由基损伤的,这些氧自由基是由以下诱变剂产生的,如:高氧张力、γ-射线、诱裂剂和产生活性氢氧根的药物。FA细胞红细胞过氧化物歧化酶(SOD)的水平降低,而白细胞SOD的浓度正常。也有红细胞SOD、过氧化氢酶和谷胱甘肽过氧化酶水平正常而谷胱甘肽转移酶水平上升的报道。在FA,成纤维细胞SOD的水平正常,Mn-SOD、过氧化氢酶和谷胱甘肽过氧化酶的浓度增加,因此有人提出氧化剂的作用可能限于造血系统。SOD或过氧化氢酶加入到FA的淋巴细胞培养中能减少断裂的数量,其他的研究者发现SOD、过氧化氢酶或半胱氨酸可减少MMC诱导的断裂。在高氧张力的情况下培养淋巴细胞导致一些FA细胞的任意断裂的数量增加,正常细胞和加入MMC后的所有FA细胞均没有增加。Clarke等研究了FA细胞的凋亡,显示在5%氧浓度时暴露在MMC中与正常细胞相同,当20%氧浓度时则对MMC超敏,暗示对FA细胞的毒性作用是MMC产生的氧反应产物(ROS)引起的,而不是DNA交联形成。FA的氧敏感性涉及控制ROS过度产生的复杂系统或耐受氧诱导损害的能力,除线粒体外,细胞内产生ROS很大程度上是由于细胞色素P450酶系统,有研究表明FANCC蛋白与NADPH细胞色素P450还原酶、FANCG蛋白与细胞色素P4502E1(CYP2E1)之间相互作用,这两种酶都产生R0S。低氧张力和抗氧化剂被用于改善生长和减少FA细胞任意或MMC诱导的染色体断裂。
(3)细胞周期调控异常:FA细胞生长缓慢,G2期延长。对类人猿病毒40(SV40)或腺病毒12的转换是敏感的并表达SV40T抗原。不管是任意的或诱变剂治疗后,FA细胞的姊妹染色单体互换一般不增加。在体外交联中FA细胞是低突变的,与正常细胞一样,主要的突变是缺失而不是点突变。FA细胞在细胞周期调控中有缺陷,G2/M期转换延迟,这进一步增加了交联剂或高氧浓度的暴露。G2/M检查点与基因组的监视和进入有丝分裂前的损伤修复有关,FA细胞存在同源性重组增加和非同源性终末连接缺陷,FA细胞不能有效的修复DNA损伤,因而停滞在G2期。G2期阻滞能被细胞周期蛋白家族中的一种SPHAR的过度表达以及咖啡因矫正,后者活化了周期依赖性激酶cdc2和废弃了一个G2的检查点。
(4)细胞凋亡和端粒维持:许多研究显示FA细胞凋亡调节异常,在一个研究中,4个FA淋巴细胞系加入MMC治疗导致了凋亡增加,但其他的研究显示FA细胞的自发凋亡增加,γ射线照射时凋亡减少,而加入MMC后凋亡没有变化。凋亡的增加可能与FA细胞修复损伤的能力有关,也可能在凋亡途径中,FA蛋白与其他蛋白相互作用的缺陷有关。FA细胞中可检测到端粒缩短加速,但在其他类型的AA和MDS中也存在。这可能是由于一个造血干细胞经过多于正常的细胞分裂次数而产生成熟细胞之缘故。最近的研究显示,除了端粒复制缩短,在FA细胞的端粒序列也有较高的断裂发生,提示端粒维持缺陷。
(5)造血缺陷:FA的造血缺陷被证明在祖细胞水平。伴AA和不伴有AA的FA患者,从骨髓CFU-GM,CFU-E,BFU-E和血BFU-E来源的克隆均减少。大多数体外细胞培养的数据以及FA患者能通过BMT治愈,提示FA的再生障碍是多能干细胞的缺陷。造血组织是对放射和细胞毒性治疗引起的DNA损伤最敏感的组织之一。其他的DNA修复障碍性疾病,如着色性干皮病或共济失调-毛细血管扩张症,发生恶性肿瘤的危险增加,但没有与FA同样高的骨髓衰竭的发生率。提示FA蛋白在造血干细胞的维持中起了特别关键的作用,并且牵涉到FA途径的不同功能的联合。引起骨髓衰竭的造血干细胞的最初的衰退,可能是由于端粒维持缺陷及高凋亡率,随后,在基因不稳定的基础上,由于细胞因子信号环境的改变,选择压力促使突变克隆的产生,导致了MDS和AML。体内及体外培养均观察到FA患者细胞因子产生的异常,血浆中肿瘤坏死因子-α(TNF-α)的水平增加,而干扰素-γ没有增加。FA淋巴细胞或成纤维细胞产生的白介素-6(IL-6)减少,加入IL-6可以修正MMC的细胞毒性。患者的淋巴细胞过度的产生TNF-α,加入IL-6减少TNF-α的过度产生,TNF-α抗体降低了对MMC的敏感性。SCF的产生正常或减低,而GM-CSF和G-CSF的产生无规律性,可减少或增高。
症状体征
本病根据再生障碍性贫血,生长迟缓、佝偻病、多尿及脱水、酸中毒、电解质紊乱等相应的临床表现;血生化检查见低血钾低血磷、低血钠、低血氯性酸中毒、高AKP、低血尿酸、糖尿而血糖正常全氨基酸尿、尿pH低而尿氨和可滴定酸低;X线检查有骨质疏松、佝偻病表现,均有助于诊断,注意询问家族史。应注意原发病的诊断,如胱氨酸储积病者,眼裂隙灯检查可见角膜有胱氨酸结晶沉着,骨髓或血白细胞中胱氨酸含量增加并见到胱氨酸结晶,对本病确切诊断十分重要由于多种类型Fanconi综合症可通过特异性治疗及对症处理取得良好疗效因此病因诊断尤为重要。
临床表现多见发育迟缓,佝偻病。由于慢性代谢性酸中毒而软弱、恶心、呕吐,由于低钾血症而致肌无力、麻痹、腹胀、便秘、心音低钝,由于肾浓缩功能差而烦渴、多尿甚而脱水。
1.原发性Fanconi综合征 是一种常染色体隐性遗传疾病,诊断时的平均年龄男性是6.5岁,女性是8岁。发病年龄范围从出生到48岁。男女比例1.2∶1。发病无种族或地区差异,一家中可有兄弟姐妹多人发病。早期患者的诊断是在再生障碍性贫血(白血病或肿瘤)发生时做出的;最近认为,患者的同胞有阳性的染色体断裂或有特征性躯体异常,就诊者即使没有贫血也可以做出FA的诊断。FA为一综合征,临床表现主要包括3方面:骨髓衰竭、各种发育异常和肿瘤易感性增加。
(1)成人型Fanconi 综合征:10~20 岁以后起病,有多种肾小管功能障碍,如全氨基酸尿、糖尿、磷酸盐尿、高血氯性酸中毒、低钾血症等。突出的症状是软骨病,少数病例可有酮症晚期可出现肾功能衰竭。
(2)婴儿型Fanconi 综合征:多于6~12 个月发病,多尿、烦渴、脱水、便秘、无力、拒食、发热,生长发育迟缓肾性氨基酸尿,可有抗维生素D 佝偻病及严重营养不良现象。实验室检查呈低血钾、低血磷、低血钙及碱性磷酸酶增高、高氯性代谢性酸中毒、尿中可滴定酸及NH4+可减少,尿糖微量或4~5g/d,血糖正常,急性起病者预后差,常死于尿毒症。慢性起病者多于2 岁以后发病,症状较轻,突出表现为侏儒和(或)抗维生素D 佝偻病。
(3)特发性刷状缘缺失型Fanconi 综合征:1984 年Manz 等首次报道1 例小儿由于近曲小管刷状缘完全缺失而引起Fanconi 综合征,因为葡萄糖及各种氨基酸载运系统完全丧失,故这些物质的清除率近于肾小球滤过率。
(4)Alter分型:根据血液学特点,Alter等于1991年把FA分为6个临床亚型: ①重型再生障碍性贫血,依赖输血,雄激素无效或未接受雄激素治疗。 ②重型再生障碍性贫血,依赖输血,正接受雄激素治疗,但疗效不佳。 ③中、重度再生障碍性贫血,不依赖输血,雄激素治疗有效。 ④中、重度再生障碍性贫血,不依赖输血,未接受雄激素治疗或雄激素治疗无效。 ⑤有骨髓衰竭的特征,如轻度贫血、粒细胞减少、血小板减少;大红细胞,HbF增高。病情稳定,不需输血和雄激素治疗。 ⑥血象正常,HbF正常或轻度异常,不需输血和雄激素治疗。
(5)anconi综合征不是全部具备上述3个特征。 ①婴儿型:也称急性型,特点有: A.起病早,6~12个月发病。 B.常因烦渴、多饮、多尿、脱水、消瘦、呕吐、便秘、无力而就诊。 C.生长迟缓、发育障碍,出现抗维生素D佝偻病及营养不良、骨质疏松甚至骨折等表现。 D.肾性全氨基酸尿,但血浆氨基酸正常。 E.低血钾,低血磷,碱性酸酶活性增高,高氯血症性代谢性酸中毒,尿中可滴定酸及NH4 可减少,尿糖微量或增多,血糖正常。 F.预后较差,可死于尿毒症性酸中毒或继发感染。 ②幼儿型:起病较晚(2岁以后),症状较婴儿型轻,以抗维生素D佝偻病及生长迟缓为最突出表现。 ③成人型:特点有: A.10~20岁或更晚发病。 B.多种肾小管功能障碍;如糖尿、全氨基酸尿、高磷酸盐尿、低血钾、高氯酸中毒。 C.软骨病往往是突出表现。 D.晚期可出现肾功能衰竭。
(6)血液学异常: FA最重要的临床特征是血液学异常,FA是遗传性骨髓衰竭综合征最常见的类型,AA、MDS和AML在纯合子的发生率都显著增加。患者出生时的血细胞计数是正常的,最早被检测到的异常是大红细胞症,随后出现血小板减少和中性粒细胞减少。40岁以前出现血液学异常的概率是98%,最常见的血液学异常是血小板减少和全血细胞减少,与骨髓增生低下有关。53%的患者以全血细胞减少发病,38%的患者以血小板减少发病,随访20年后有84%的患者发展为全血细胞减少,少部分患者以贫血、中性粒细胞减少发病,部分患者发病时即为骨髓增生异常综合征(MDS)或急性白血病(尤其是急性髓系白血病)。临床上,FA患者有出血、苍白和(或)反复感染。
(7)其他表现:身材矮小的FA患者28例检测了生长激素(GH)水平,有22例GH缺乏,GH激素治疗15例患者有12例长高,无一例患者造血改善,GH治疗的患者中一例死于急性髓系白血病。最近报道44%的患者有GH缺乏,36%的患者有甲状腺功能减低。重组GH在生长和造血中的作用还需要进一步观察,而且GH治疗和白血病之间的可能联系需要仔细探讨。
2.继发性Fanconi综合征 因病因不同,表现有所不同。 本病根据再生障碍性贫血,生长迟缓、佝偻病、多尿及脱水、酸中毒、电解质紊乱等相应的临床表现;血生化检查见低血钾、低血磷、低血钠、低血氯性酸中毒、高AKP、低血尿酸、糖尿而血糖正常,全氨基酸尿、尿pH低而尿氨和可滴定酸低;X线检查有骨质疏松、佝偻病表现,均有助于诊断,注意询问家族史。应注意原发病的诊断,如胱氨酸储积病者,眼裂隙灯检查可见角膜有胱氨酸结晶沉着,骨髓或血白细胞中胱氨酸含量增加并见到胱氨酸结晶,对本病确切诊断十分重要。由于多种类型Fanconi综合征可通过特异性治疗及对症处理取得良好疗效,因此病因诊断尤为重要。
(1) 胱氨酸储积症:本症又称Lignac-Fanconi 综合征,系胱氨酸沉着于细胞溶酶体而表现为Fanconi 综合征。正常人细胞内溶酶体是细胞内蛋白降解的部位,细胞内蛋白降解产生氨基酸通过溶酶体膜转输系统输入胞质而被再利用。本病因溶酶体内胱氨酸运载体有缺陷,使胱氨酸在溶酶体中储积,从而破坏了溶酶体的完整性,并可使具有破坏性的溶酶体酶漏至细胞浆,影响了功能。本病与胱氨酸尿症不同,后者是肾小管上皮转运胱氨酸障碍,只引起胱氨酸尿,前者则引起许多器官细胞内胱氨酸储积,肾脏是主要受累器官之一。胱氨酸储积病所引起的Fanconi 综合征不同于其他原因所致Fanconi 综合征,常以失钾、脱水、多饮、渗透性利尿为突出表现。临床上可分3 型:
(2) ①婴儿型或肾病型:胱氨酸沉积于各种组织溶酶体内,在白细胞内可能比正常大80 倍,肾脏髓质可能沉着近100 倍,因肾小管损伤出现各种症状,患儿多于6 个月左右开始发病,多尿、烦渴、便秘、多饮、呕吐、拒食、消瘦、发育障碍,由于脱水有反复发热,可发生维生素D 缺乏病及侏儒症。由于角膜、结膜的胱氨酸沉着而畏光,眼底周围色素脱失,可引起末梢视网膜病变。此外尚可表现为甲状腺功能低下、糖尿病、脾大、脑水肿、肌病等。肾小管功能障碍表现为肾浓缩功能障碍、氢离子排泄功能障碍,而至尿液不能酸化至pH 5.5 以下,呈肾小管性酸中毒。
②儿童型或中间型:10 岁左右发病,进展较慢,骨病不严重,无侏儒症。组织胱氨酸含量远较婴儿型为低,白细胞内胱氨酸含量为正常的30 倍,也可表现为肾脏病变,甚至发展为尿毒症,骨骼畸形、畏光、视网膜病变、胱氨酸引起的脾大也可产生。Fanconi 综合征表现不明显。
③成人型:无肾病表现,以其他器官功能障碍为主。成人型又可分为急性与慢性,前者与婴儿型类似,后者与儿童型类似。
通过骨髓片、白细胞、直肠黏膜中的结晶分析或裂隙灯检查角膜有胱氨酸结晶而诊断。婴儿型因患儿拒食引起饥饿性酮症加上肾小管性糖尿,易误诊为幼儿糖尿病,需提高警惕。
(2)Lowe 综合征:本综合征系Lowe 1952 年首先报道,亦称眼-脑-肾综合征,临床表现特点为:
①眼症状:先天性白内障(双侧)伴有先天性青光眼(牛眼)、视力严重障碍、眼球震颤及畏光。
②脑症状:严重智力发育迟缓,肌张力低、腱反射减弱或消失,患儿常哭泣样尖叫。
(3)肝豆状核变性(Wilson 病):本病系少见的隐性遗传性代谢性疾病。因为血浆铜蓝蛋白(ceruloplasmin)含量降低,铜氧化酶活性降低导致肠道大量吸收铜,大量铜沉积于肝、脑、角膜、肾小管引起相应的症状。铜沉积于脑及肝引起锥体外系神经症状及肝硬化,铜沉积于角膜引起Kayser-Fleischer 环。铜沉积于近端肾小管及远程肾小管引起Fanconi 综合征,可伴有重碳酸盐丢失和肾钙质沉着伴高钙尿症,肾小管性酸中毒,肾钙化,肾结石。本病可用青霉胺治疗,促进铜从尿中排出但停用后会复发。其他治疗如二巯丙醇(BAL)可增加铜排出,口服硫化钠可改善神经系统和肝症状,但肾小管病变无改善,骨化醇可治疗骨病变。
小儿范科尼综合征吃什么好
鉴别诊断
1.应与肾小管酸中毒相区别。
2.进行病因诊断遗传性见于原发性遗传性范可尼综合征;也继发于胱氨酸病、肝豆状核变性、眼脑肾综合征(综合征)、果糖不耐受、酪氨酸血症、半乳糖血症、维生素依赖性佝偻病等。.获得性多发性骨髓瘤、淀粉样变、干燥综合征、肾病综合征、肾移植、维生素缺乏、甲状旁腺功能亢进、婴儿肾静脉栓塞、肝肿瘤、钾缺乏、间质性肾炎(有抗肾小管基膜抗体)等。.中毒重金属如汞、铅、铜、镉、铀等,化学物质有来苏、水杨酸盐、硝基苯、马来酸、甲基--铬、过期四环素等。
3.血小板减少症-桡骨缺失综合征(TAR) 系常染色体隐性遗传性疾病。它除有先天性增殖障碍性血小板减少症外,两侧桡骨缺失是其基本特征,因此可以应用胎儿放射照相术进行诊断。在出生时或新生儿期出现血小板减少伴有桡骨缺失,但双侧拇指存在,没有血液系统恶性肿瘤或实体瘤增加的危险,与FA不同。
4.VATER/VACTERL(vertebral defects,anal atresia,tracheoesophageal fistula, renal defects and radial limb defects) 患者有脊椎缺损、肝门闭锁、气管食管瘘、桡骨及肾脏发育异常,与FA的临床表现有重叠。
5.先天性纯红细胞再生障碍性贫血(DBA) 是以红系祖细胞成熟缺陷为特征,是一种少见的先天性纯红细胞再生障碍,以贫血为主要临床表现,并累及多系统组织为本病主要临床特征。1936年由Joseph报道一种小儿纯红再障,认为是先天性的或者是遗传性的。1岁以内出现正细胞或大细胞性贫血,1/3以上的患者有先天畸形,常累及头部(小颌,唇裂)、上肢和泌尿生殖系统。大多数患者为散发,少数为常染色体显性或隐性遗传。以上3种疾病的DEB/MMC试验无染色体断裂增加,可与FA鉴别。
6. Nijmegen断裂综合征 是一种罕见的常染色体隐性遗传性疾病,表现为高辐射敏感性、免疫缺陷、发育不良、早衰、肿瘤易感性、小头畸形等.现就NBS症状。由NBS1突变引起,以免疫缺陷、小头畸形和对电离辐射超敏为特征,大多数患者是南斯拉夫人。最近的一个研究中,8个俄国的NBS患者,3个有造血异常或AML,因此,临床上与FA非常相似。如果诊断困难可以考虑进行NBS突变检查。表1为各种不同病因下Fanconi综合征的特点和区别。
并发症
1.并发症 常并发发育障碍、抗维生素D佝偻病及营养不良、骨质疏松甚至骨折、肾小管性酸中毒、低钾血症、继发性甲状旁腺功能亢进、肾性骨病、骨畸形、骨软化症;肾结石、低血磷、高氯血症,死于尿毒症性酸中毒或继发感染。
抗维生素 D 性佝偻病是一种肾小管遗传缺陷性疾病,该病主要是 由于 位于 X 染色体上的 PHEX 基因的突变,导致 肾小管回吸收磷减少所致。肠道吸收钙、磷不良,血磷降低,一般 在 0.65 ~ 0.97/mmol/L(2 ~ 3mg/dl) 之间 ,钙磷乘积多在 30 以下,骨质不易钙化。
2.多伴发先天性畸形 先天性畸形是指出生前在母体内已形成的外形或体内有可识别的结构和功能缺陷(不包括代谢疾病)。据统计近20年来,新生儿出生有缺陷者的发病率有逐渐增高趋势,现为3%-5%。其产生原因为遗传性和环境因素两大类。前者是指从父母处接受的遗传达室物质所致,即染色体异常和单基因、多基因遗传.后者是指在胎儿发育过程中,受外界因素或母体变化的影响而致畸。
在婴儿期诊断的FA患者有较高的比例有特征性躯体异常,如畸形包括拇指和桡骨、肾脏、头、眼睛和耳朵以及胃肠道畸形。相反,在年长儿诊断的FA患者,躯体异常少见。身材矮小和皮肤色素问题随着年龄增加而显现出来。FA患者的典型异常包括:身材矮小,拇指或桡骨不发育或缺如或多指,小眼球,小头畸形,皮肤色素沉着、浅褐色斑和色素脱失斑。以及特征性面部表现如鼻基底部增宽,眦上皱襞和小颌。大约1/3的患者有肾脏异常,包括肾发育不全,马蹄形肾或双输尿管。生殖系统发育不全(女性月经来潮较晚,月经不规则,绝经早,妇科肿瘤多;男性性腺、尿道发育不完全)。少见的异常包括胃肠道、心脏和中枢神经系统等部位的缺陷。只有血象和骨髓象异常的而无先天畸形的患者称为Estren-Dameshek综合征,这些患者仅代表FA范畴中的一部分。
3.肿瘤易感性 FA患者发生白血病和肿瘤的报道多于200例,总的发生率大于15%。急性白血病占100例以上,占FA患者的10%,主要是急性髓系白血病。发生MDS的危险是10%~35%。有克隆性细胞遗传学异常的患者发生MDS或AML的危险明显高于染色体核型正常者。克隆性细胞遗传学异常累及染色体1和7,其他的异常包括染色体部分或完全的缺失、易位和标记染色体。FA患者发生实体瘤的危险性随年龄的增大而增加,大多数是鳞状细胞癌,最常累及消化道和女性生殖系统。
4.FA异质性 在FA患者的亲戚中可发现先天性畸形、泌尿生殖系统和手的特殊异常。一些FA患者的父母亲有身体畸形如身材矮小,但没有血液学疾病。Pefidou和Barrett注意到有关的异质性有HbF水平的增加,自然杀伤细胞数量的减少和对丝裂霉素反应差。FA的异质性也可以通过染色体断裂分析被检测。
预防保健
暂无相关资料
治疗用药
本病是遗传性疾病者,对其发病无特效预防办法,对继发性或已确诊本病的患者应积极对症治疗,以预防并发症的发生和延缓肾衰竭。同此,应重视孕期保健,积极防治各种感染,尤其是病毒感染性疾病,避免理化因素、毒性物质的损害。
保健品查询热门体检套餐





大家都在看
- 乳腺癌
乳腺癌(mammarycancer)是女性最常见的恶性肿瘤之一,发病率占全身各种...[详细]
- 男性不育症
男性不育症是指由于男性因素引起的不育。一般把婚后同居2年以上未采取任何避孕措施而...[详细]
- 不孕症

不孕症(infertility)系指凡婚后夫妇有正常的性生活、未避孕、同居2年而...[详细]
- 早泄
早泄是指阴茎插入阴道后,在女性尚未达到性高潮,而男性的性交时间短于2分钟,提早射...[详细]
- 胃癌

胃癌是我国最常见的恶性肿瘤之一,在我国其发病率居各类肿瘤的首位,每年约有17万人...[详细]
- 前列腺炎
前列腺炎是指前列腺特异性和非特异感染所致的急慢性炎症,从而引起的全身或局部症状。...[详细]
- 流产
流产(abortion)为妇科常见疾病,如处理不当或处理不及时,可能遗留生殖器官...[详细]
- 乙肝
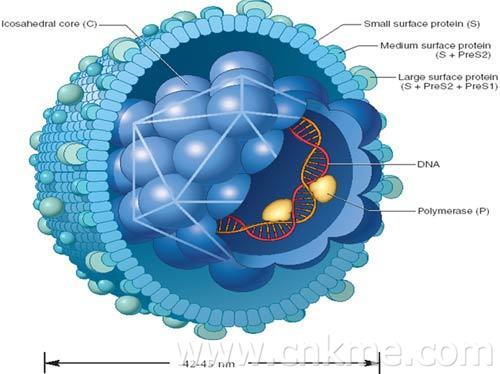
乙型病毒性肝炎是由乙型肝炎病毒(HBV)引起的一种世界性疾病。发展中国家发病率高...[详细]
- 颈椎病
颈椎病又称颈椎综合征,是颈椎骨关节炎、增生性颈椎炎、颈神经根综合征、颈椎间盘脱出...[详细]
- 心绞痛
心绞痛(anginapectoris)是冠状动脉供血不足,心肌急剧的、暂时缺血与...[详细]
最新
- 肿瘤

肿瘤(tumour)是指机体在各种致瘤因子作用下,局部组织细胞增生所形成的新生物...[详细]
- 心脏病

心脏病是一类比较常见的循环系统疾病。循环系统由心脏、血管和调节血液循环的神经体液...[详细]
- 脑溢血
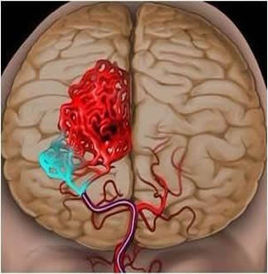
脑出血是指非外伤性脑实质内血管破裂引起的出血,占全部脑卒中的20%~30%,发生...[详细]
- 内分泌性高血压
内分泌组织增生或肿瘤所致的多种内分泌疾病,由于其相应激素如醛固酮、儿茶酚胺、皮质...[详细]
- 寒痹
寒痹是怎么回事?寒痹,病名。一名痛痹、骨痹。指寒邪偏重的痹证。《灵枢•贼风》:“...[详细]
- 小儿46-XY单纯性腺发育不全综合征
46-XY单纯性腺发育不全综合征(simple46,XYgonadaldigen...[详细]
- 小儿情感交叉擦腿综合征

情感交叉擦腿综合征(masturbationsyndrome)是一个病因不明,治...[详细]
- 小儿先天性肌强直综合征

肌强直综合征(myotonicsyndrome)是一组较为少见的遗传性疾病,包括...[详细]
- 新生儿单纯疱疹病毒感染
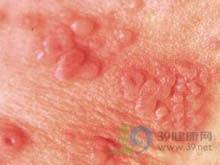
新生儿单纯疱疹病毒感染(herpessimplexviralinfectiono...[详细]
- 小儿猫叫综合征

猫叫综合征(catscrysyndrome)是由于第5号染色体短臂缺失(5p缺失...[详细]

