脊髓性肌萎缩
更新时间:2026-04-28
脊髓性肌萎缩(spinal muscular atrophy,SMA)系指一类由于以脊髓前角细胞为主的变性导致肌无力和肌萎缩的疾病。首先由Werdnig(1891)和Hoffmann(1893)报道,故又称Werdnig-Hoffmann病。根据起病年龄和病变程度可将本病分为4型:Ⅰ~Ⅲ型称为儿童型SMA,属于常染色体隐性遗传病,其群体发病率为1/6000~1/10000,是婴儿期最常见的致死性遗传病。20~30岁以上起病的SMA,归为第Ⅳ型,可呈常染色体隐性、显性和X连锁隐性等不同遗传方式,其群体发病率约为0.32/10000。
由于存在各种临床和遗传学方面的特点,故目前普遍认为本病应从运动神经元疾病中分出,成为一组独立疾病。
病因
(一)发病原因
Ⅰ~Ⅲ型属于常染色体隐性遗传病,是婴儿期最常见的致死性遗传病。第Ⅳ型为常染色体隐性、显性和X连锁隐性等不同遗传方式。
(二)发病机制
SMA的病因和发病机制一直是神经病学研究中的一个难题。近几年来在SMA基因定位的研究方面取得了很大进展。1995年,不同研究小组分别报道了3个SMA候选基因。法国Lefebvre等在5q13.1区域发现了运动神经元生存(survival motor neuron,SMN)基因,全长约20kb,含8个外显子,其转录产物约1.7kb,编码294个氨基酸,功能未知。在一条染色体上该基因具有两个拷贝,二者间有5个碱基的差别,在端粒侧称SMNt,着丝粒侧称SMNc。研究表明,SMNt第7、8号外显子在98.6%SMA患者中纯合缺失或截断,另1.4%患者有小缺失或点突变,这强烈支持SMN是SMA重要的决定基因。随后Roy等在5q13区域另克隆到神经细胞凋亡抑制蛋白(neuronal apoptosis inhibitory protein,NAIP)基因,有16个外显子,全长70kb,编码1232个氨基酸。45%SMA-Ⅰ型和18%SMAⅡ、Ⅲ型患者存在NAIP基因第5、6号外显子缺失,而2%的正常对照亦缺失5、6号外显子,提示NAIP基因亦与SMA协发病相关。至于成年型SMA,仅部分发现有SMN基因的缺失,提示与儿童型SMA有相似的基因改变,但大部分患者的基因定位尚未确定,发病机制未明。
病理变化主要位于脊髓前角,其运动细胞明显减少,呈退行性变,残留的神经细胞呈固缩、核溶解,脊髓前根轴突变细,轴突外周细胞肿胀。脑干运动神经核变性,以面神经、迷走神经、舌下神经多见。肌肉病理检查见下述辅助检查部分。
症状体征
1.SMA-Ⅰ型 亦称Werdnig-Hoffmann病。约1/3病例在宫内发病,其母亲可注意到胎动变弱。半数在出生1个月内起病,几乎所有病例均在5个月内发病。发病率约为1/10000出生婴儿,男女发病相等。多于出生后不久即表现肌张力低下,肌无力以四肢近端肌群受累为主,躯干肌亦无力。患儿吸吮及吞咽力弱,哭声低微,呼吸浅,可出现胸廓反常活动。翻身及抬头困难。腱反射消失。触诊可发现四肢肌萎缩,但常被皮下脂肪掩盖。眼球运动正常。括约肌功能正常。可见舌肌萎缩和束颤。10%病例可有关节畸形或挛缩。本型预后差。约95%死于出生后18个月。
2.SMA-Ⅱ型 发病较SMA-Ⅰ型稍迟,通常于1岁内起病,极少于1~2岁起病。发病率与SMA-Ⅰ型相似。婴儿早期生长正常,但6个月以后运动发育迟缓,虽然能坐,但独站及行走均未达到正常水平。1/3以上患儿不能行走。20%~40%患儿10岁以前仍具行走能力。多数病例表现严重肢体近端肌无力,下肢重于上肢,而呼吸肌、吞咽肌一般不受累。有1/3病例面肌受累。50%以上病例可见舌肌及其他肌肉纤颤。腱反射减弱或消失。本型具有相对良性的病程,多数可活到儿童期,个别活到成年。
3.SMA-Ⅲ型 又称Kugelberg-Welander病。一般于幼儿期至青春期起病,而多数于5岁前起病。起病隐袭,表现为进行性肢体近端肌无力和萎缩。早期大腿及髋部肌无力较显著,以致病孩行走呈鸭步,登梯困难,逐渐累及肩胛带及上肢肌群。脑神经支配的肌群通常未受累及,但可出现面肌、软腭肌无力。眼外肌正常。约1/4病例伴发腓肠肌假性肥大,此几乎均见于男性患者。半数患者早期可见肌束颤动。弓形足亦可见到。腱反射减弱或消失。感觉正常。本型预后良好,尤其是女性患者。生存期通常能达到成年,许多患者能有正常寿命。表现较严重病例往往为男性患者。本型血清CPK可有不同程度增高。EMG除呈神经源性改变外尚可与肌源性损害混杂存在,因此须注意与肌营养不良症鉴别。
4.SMA-Ⅳ型 统称成年型SMA。发病年龄为15~60岁,多见于35岁左右。起病和进展均较隐袭,但亦有呈进行性加重或相对静止的病例报道。本型预后相对良好,行走能力常可保持终生。发病率小于0.5/10万。本型中约1/3病例呈常染色体显性遗传,表现为近端肌无力,进展速度稍快,约5年后丧失跑步能力。尚有常染色体隐性遗传类型,则一般表现更加良性病程。另一种类型为X-连锁隐性遗传,又称脊髓脑干型SMA(Kennedy病),其发病年龄不等,但常于40岁前发病。早期表现痛性肌痉挛,可先于肌无力前数年出现。近端肌无力常从下肢开始,逐渐波及肩胛带肌、面肌及延髓支配诸肌。下面肌及舌肌可见束颤。数年后可出现吞咽困难及呐吃。约50%病例合并一些内分泌功能障碍,表现男性乳房女性化及原发性睾丸病变。
5.其他类型SMA
(1)远端型SMA:本型约占SMA的10%,为常染色体显性或隐性遗传形式。前者于20岁前发病,后者稍迟,且症状较轻。多数患者表现缓慢进展的下肢远端肌无力和萎缩,胫骨前肌和腓骨肌群尤易受累。弓形足和脊柱侧弯亦较常见。约半数病例上肢远端迟早也会受累,但程度较轻。无感觉障碍。周围神经传导速度正常。
(2)慢性不对称型SMA:本型于16~45岁起病,男性患者为女性患者的两倍。表现一个或多个肢体不对称性肌萎缩,而无锥体束或延髓受累。肌无力可以近端或远端为主,起病时相对局限于单个肢体。本型自然病程较长,甚至超过30年。
(3)肩胛腓型SMA:发病年龄30~40岁。表现肩胛带肌及下肢远端肌(尤其是腓肠肌)明显无力和萎缩。弓形足也较常见。
(4)单肢型SMA:日本和印度曾报道一些病例,其发病年龄各异,男性多见。起病相对较快,而后进入非进展期。由于局限性前角细胞受损,多表现单臂明显肌萎缩。EMG显示严格限制于单个肢体的异常。延髓肌及其他肌肉不受侵犯。日本文献中称青年型单肢SMA为平山(Hirayama)病。
(5)此外尚有延髓SMA并发耳聋(Viatetto-Vanlaere综合征)、儿童延髓型SMA(Fazio-Londe综合征)、眼咽型SMA、面肩肱型SMA、氨基已糖苷酯酶A缺陷等少见类型。
根据本病仅累及下运动神经元,四肢呈进行性弛缓性瘫痪,近端重于远端,下肢重于上肢等临床表现,结合颈椎或腰椎影像学未见与临床相一致的表现,以及肌电图、肌肉病理检查等特点,一般不难做出诊断。
如有阳性家族史则更支持诊断。基因检测可为确立诊断提供可靠的证据。依据临床特点、发病年龄、预后和遗传方式等再做出分型诊断。
脊髓性肌萎缩吃什么好
鉴别诊断
一般在本病早期或不典型病例,应注意与下列疾病鉴别:
1.新生儿型重症肌无力 其母均为重症肌无力患者,此与母亲血液中抗Ach受体抗体通过胎盘到达胎儿体内有关。一般于出生后即表现吸吮困难、哭声无力、四肢运动减少等症状。多数患儿于2~6周内症状逐渐好转,且用胆碱酯酶抑制剂治疗有效。
2.先天性肌张力不全(Oppenheim病) 出生后出现肌张力低下,未见肌肉萎缩,肌电图及肌肉活检均无异常。
3.进行性肌营养不良 一般在SMA-Ⅱ、Ⅲ型患儿中需注意与Duchenne型或Becker型肌营养不良进行鉴别。后者几乎均有假性肥大征象,其血清CPK极高,特别在病程的早期阶段,EMG和肌肉活检均呈肌源性损害,故一般鉴别并不困难。SMA-Ⅳ型易与肢带型肌营养不良和多发性肌炎等混淆,但从临床表现、血清酶学、EMG以及肌肉活检等方面的特点分析,也不难区别。
并发症
不同类型SMA的症状体征可以是本病表现,也可以看作本病并发症(参见上述临床表现)。另外,应注意继发的肺部感染、尿路感染、褥疮等。
预防保健
防止患儿出生是预防本病的最有效方法。新近国内有学者联合应用PCR-SSCP、PCR-限制性酶切及连锁分析法进行SMA的产前基因诊断,其准确率和成功率较高,值得进一步推广和应用。
治疗用药
(一)治疗
目前尚无治疗本病的特效方法。支持和对症治疗是本病的主要疗法。应加强营养,注意提高机体抵抗力,积极预防呼吸道感染。可配合理疗、针灸、按摩以及被动运动等方法,进行运动功能锻炼并防止肢体挛缩。近年来,国外曾有应用神经干细胞治疗SMA的动物实验报道,根据他们的观察结果表明,移植的干细胞可移行至受损的神经元区域,部分已分化为神经元。下一步将逐步进入临床试验,这将给本病的治疗带来新的希望。
(二)预后
本病多数预后不良。SMA-Ⅰ型预后最差,约95%死于出生后18个月。SMA-Ⅱ型具有相对良性的病程,多数可活到儿童期,个别活到成年。SMA-Ⅲ型预后良好,尤其是女性患者。生存期通常能达到成年,许多患者能有正常寿命。表现较严重病例往往为男性患者。SMA-Ⅳ型预后相对良好,行走能力常可保持终生。慢性不对称型SMA自然病程较长,甚至超过30年。
保健品查询热门体检套餐





大家都在看
- 乳腺癌
乳腺癌(mammarycancer)是女性最常见的恶性肿瘤之一,发病率占全身各种...[详细]
- 男性不育症
男性不育症是指由于男性因素引起的不育。一般把婚后同居2年以上未采取任何避孕措施而...[详细]
- 不孕症

不孕症(infertility)系指凡婚后夫妇有正常的性生活、未避孕、同居2年而...[详细]
- 早泄
早泄是指阴茎插入阴道后,在女性尚未达到性高潮,而男性的性交时间短于2分钟,提早射...[详细]
- 胃癌

胃癌是我国最常见的恶性肿瘤之一,在我国其发病率居各类肿瘤的首位,每年约有17万人...[详细]
- 前列腺炎
前列腺炎是指前列腺特异性和非特异感染所致的急慢性炎症,从而引起的全身或局部症状。...[详细]
- 流产
流产(abortion)为妇科常见疾病,如处理不当或处理不及时,可能遗留生殖器官...[详细]
- 乙肝
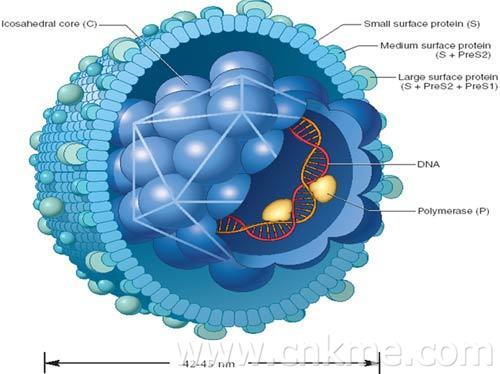
乙型病毒性肝炎是由乙型肝炎病毒(HBV)引起的一种世界性疾病。发展中国家发病率高...[详细]
- 颈椎病
颈椎病又称颈椎综合征,是颈椎骨关节炎、增生性颈椎炎、颈神经根综合征、颈椎间盘脱出...[详细]
- 心绞痛
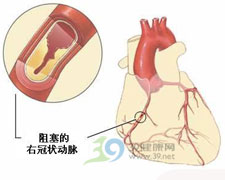
心绞痛(anginapectoris)是冠状动脉供血不足,心肌急剧的、暂时缺血与...[详细]
最新
- 肿瘤

肿瘤(tumour)是指机体在各种致瘤因子作用下,局部组织细胞增生所形成的新生物...[详细]
- 心脏病

心脏病是一类比较常见的循环系统疾病。循环系统由心脏、血管和调节血液循环的神经体液...[详细]
- 脑溢血
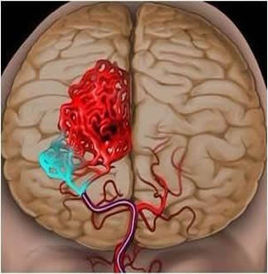
脑出血是指非外伤性脑实质内血管破裂引起的出血,占全部脑卒中的20%~30%,发生...[详细]
- 内分泌性高血压
内分泌组织增生或肿瘤所致的多种内分泌疾病,由于其相应激素如醛固酮、儿茶酚胺、皮质...[详细]
- 寒痹
寒痹是怎么回事?寒痹,病名。一名痛痹、骨痹。指寒邪偏重的痹证。《灵枢•贼风》:“...[详细]
- 小儿46-XY单纯性腺发育不全综合征
46-XY单纯性腺发育不全综合征(simple46,XYgonadaldigen...[详细]
- 小儿情感交叉擦腿综合征

情感交叉擦腿综合征(masturbationsyndrome)是一个病因不明,治...[详细]
- 小儿先天性肌强直综合征

肌强直综合征(myotonicsyndrome)是一组较为少见的遗传性疾病,包括...[详细]
- 新生儿单纯疱疹病毒感染
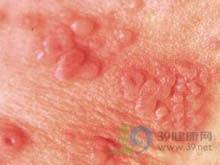
新生儿单纯疱疹病毒感染(herpessimplexviralinfectiono...[详细]
- 小儿猫叫综合征

猫叫综合征(catscrysyndrome)是由于第5号染色体短臂缺失(5p缺失...[详细]

